
Understanding how uranium dioxide (UO2) corrodes in the presence of air and water is important for several reasons. UO2 is the main component of fuel for nuclear reactors and the most economically significant uranium mineral. In addition, some bioremediation efforts to immobilize uranium at contaminated sites rely on microbes that produce uranium dioxide by chemically altering the oxidation state of other forms of uranium. In all these contexts, further oxidation of UO2 has potentially damaging consequences: It can degrade the mechanical stability of fuel rods and allow uranium compounds to seep into the environment. Researchers using the U.S. Department of Energy’s Advanced Photon Source (APS, an Office of Science user facility), have combined experimental and theoretical techniques to investigate exactly how oxygen atoms infiltrate a UO2 crystal through an exposed surface. They find that the atoms do not diffuse randomly but move to preferred interstitial locations in the lattice, causing structural changes that alter the mineral’s chemical and physical properties. The results of the study provide insight into material failure as well as conditions that help or hinder bioremediation.
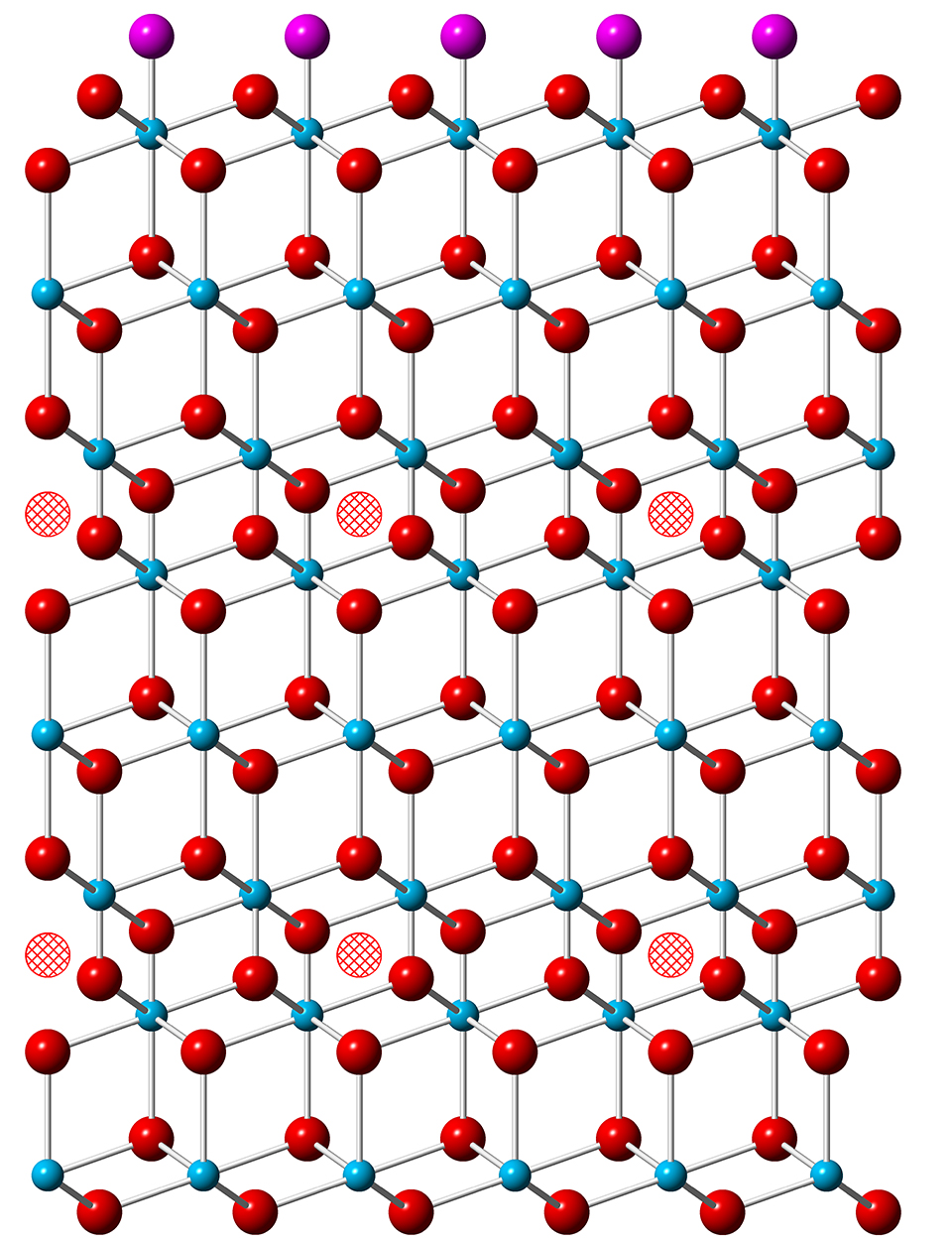
The uranium atoms in UO2 are all in the U(IV) oxidation state. The mineral can absorb interstitially up to one additional oxygen for each eight in the lattice – creating UO2.25 – with little structural change, but with some of the uranium atoms moving to higher oxidation states. Further addition of oxygen, however, leads to the appearance of U3O8, which has a larger lattice volume than UO2. This lattice rearrangement damages the integrity of the mineral and leads to material failure. In addition, uranium in the U(VI) oxidation state dissolves in water, allowing it to leach into the environment.
Although this sequence of changes is well known, details of how the lattice accommodates interstitial oxygen and how the oxidation state of uranium changes in response are not well understood, particularly when it comes to surface corrosion at ambient pressure and temperature. As a result, it has been difficult to model and predict the degradation of UO2.
Working at the GeoSoilEnviroCARS beamlines 13-BM-C and 13-ID-C at the Argonne APS, the team of researchers from The University of Chicago, the Pacific Northwest National Laboratory (PNNL), and the Stanford Synchrotron Radiation Lightsource used crystal truncation rod (CTR) x-ray diffraction to monitor changes in the surface structure of UO2 exposed to oxygen. They took a single crystal of UO2, 3-millimeters across and 0.5-millimeters thick, and polished it to expose an atomically smooth surface in the (111) plane, a natural cleavage surface. Over a period of 21 days, the crystal was exposed to dry oxygen at one atmosphere of pressure between measurements.
Before exposure to oxygen, CTR scans of UO2 revealed a surface very similar to the bulk lattice structure. As oxygen was absorbed during the experiments, the researchers detected asymmetries in the Bragg peaks as well as additional peaks at intervening positions. These changes indicated that layers of atoms just below the surface were getting closer together and that a superlattice structure, on a scale larger than the unit cell of UO2, was emerging.
Because uranium atoms scatter x-rays much more strongly than oxygen atoms, the CTR results by themselves were not able to locate exactly where in the lattice interstitial oxygens resided. The team turned to density functional theory to calculate how the electron orbitals of UO2 change for different occupancy fractions of interstitial oxygen and thereby find the lowest-energy positions for the oxygen atoms. They could then find the best match between the calculated lattice distortions and structural changes implied by the CTR scans.
In addition, the researchers performed x-ray photoelectron spectroscopy measurements at PNNL’s Environmental Molecular Sciences Laboratory to determine the relative fraction of uranium atoms in different oxidation states. This information too was compared to the results of the density functional calculations.

These investigations show that after oxygens have initially adsorbed onto the surface of UO2, the most stable location for an oxygen that penetrates into the lattice is in the third layer down. As those occupancy sites fill up, additional oxygens begin to occupy the sixth layer. Each interstitial oxygen draws some electron density from the 38 nearest uranium atoms around it, limiting the maximum number of interstitials in a layer to about 25% of the available locations. This occupancy figure is consistent with the lattice distortion revealed by the CTR measurements.
The researchers conclude that oxygen does not penetrate UO2 in the manner of a typically diffusive process. Instead, an oxidation front advances into the material in discrete steps. This new understanding of the dynamics of oxidation may lead to better predictive models for the progress of bioremediation efforts and the stability of the resulting mineral formations, the researchers say.— David Lindley
See: Joanne E. Stubbs1*, Anne M. Chaka2, Eugene S. Ilton2, Craig A. Biwer1‡, Mark H. Engelhard2, John R. Bargar3, and Peter J. Eng1, "UO2 Oxidative Corrosion by Nonclassical Diffusion," Phys. Rev. Lett. 114, 246103 (2015). DOI: 10.1103/PhysRevLett.114.246103
Author affiliations: 1University of Chicago, 2Pacific Northwest National Laboratory, 3Stanford Synchrotron Radiation Lightsource, ‡Present address: University of Michigan
Correspondence: * [email protected]
A. M. C. and E. S. I. were supported by the Geosciences Research Program at PNNL, U.S. Department of Energy (DOE) Office of Science-Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences. Support was provided by DOE Biological and Environmental Research (BER), Subsurface Biogeochemical Research, through the SLAC SFA program (DOE Contract No. DEAC02- 76SF00515). XPS data were collected in the Radiochemistry Annex at EMSL, and a portion of the DFT study was performed using the computational resources of EMSL, a national scientific user facility sponsored by DOE-BER and located at PNNL. GeoSoilEnviroCARS is supported by the National Science Foundation-Earth Sciences (EAR-1128799) and DOE-GeoSciences (DE-FG02-94ER14466). This research used resources of the Advanced Photon Source, a U.S. DOE Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Argonne National Laboratory is supported by the Office of Science of the U.S. Department of Energy. The Office of Science is the single largest supporter of basic research in the physical sciences in the United States, and is working to address some of the most pressing challenges of our time. For more information, please visit science.energy.gov.