
The chemical reactivity of molecules in solution critically depends on a complex interplay among intramolecular processes and interactions with the caging salvation shell, which surrounds a solute molecule. Accordingly, the influence of solvation on the reactivity of chemical and biological molecular species has been the subject of increasingly intense research. Ultrafast time-resolved x-ray measurement techniques that combine picosecond lasers and short-pulse x-rays in laser pump/x-ray probe experiments are powerful tools for studying this interplay in photo-active molecular systems. In a typical laser pump/x-ray probe experiment, a laser pulse excites a molecular sample to create a transient state; an x-ray pulse follows to probe the transient state at a fixed time delay relative to the laser pulse. X-ray spectroscopy and/or scattering signals can then be accumulated over many thousand such pump/probe cycles to satisfy signal-to-noise requirements. Doing so at a series of different time delays then reveals the temporal evolution of the transient state(s).
Such time-resolved laser pump/x-ray probe experiments have been carried out at synchrotrons for several years; however, few have been able to make use of the full x-ray flux available, often because of the low (kHz) repetition rate of amplified laser systems. Thanks to implementation of a high-repetition-rate (54 kHz–6.5 MHz), high-power (>10 W) laser system at the X-ray Science Division 7-ID-D beamline at the Advanced Photon Source (APS), it has become possible to fully match the repetition rate of the laser to the 6.5-MHz rate of the x-rays, and thus to more efficiently use the flux provided by the APS.
This has enabled laser pump/x-ray probe measurements incorporating simultaneous x-ray spectroscopy and x-ray scattering techniques to study light-induced intramolecular processes and solvent interactions in challenging molecular systems. It is accomplished by simultaneous, time-resolved utilization of several complementary analytical techniques that all take advantage of the thousand-fold increase in useful x-ray flux provided by the MHz scheme.
The superior signal statistics permitted the extraction and analysis of ultrafast solute-solvent dynamics for the first time, thanks to the simultaneous inclusion of both x-ray spectroscopy and x-ray scattering tools into a single experimental setup. This facilitated the analysis of otherwise quite complex data by using complementary information extracted from each tool as a vital cross-check of the analysis.
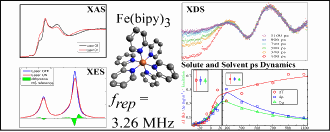
Researchers from the Technical University of Denmark, the Hungarian Academy of Sciences, the European XFEL at DESY (Germany), Argonne National Laboratory, the University of Copenhagen, SLAC National Accelerator Laboratory, and Lund University (Sweden) designed such an experiment for a highly detailed study of the photoinduced low-spin (LS) to high-spin (HS) conversion of iron(II)-tris(2,2′-bipyridine), [Fe(bpy)3]2+, in aqueous solution, to provide information on the interplay between intramolecular dynamics and the caging solvent response on a 100-ps time scale.
The setup allowed simultaneous acquisition of x-ray emission spectroscopy (XES), x-ray absorption near edge spectroscopy (XANES), and x-ray diffuse scattering (XDS) data. These complementary spectroscopic and scattering techniques are sensitive to, respectively, the spin state and configuration of the electron system, the electronic and local structure around the absorbing atom, and the global molecular structure, including the solvation shell. The combined setup thus allowed simultaneous characterization of both the electronic and geometric degrees of freedom, while making full use of the intensity of the APS for a time-resolved experiment.
By combining information from the photon-hungry techniques of XES and XDS, the excitation fraction, as well as the temperature and density changes of the solvent, were closely followed on the sub-nanosecond time scale of the HS lifetime, allowing the detection of an ultrafast change in bulk solvent density (Fig. 1).
While initially surprising, this ultrafast bulk-density increase was shown to be consistent with recent theoretical predictions from high-level molecular dynamics simulations.
Tight focusing of both the laser (<100-µm) and x-ray (<10-µm) beams allowed the researchers to obtain high excitation fractions (30–40% HS [Fe(bpy)3]2+) in the probed sample volume. The 3.26-MHz repetition rate of the experiment, the highest reported yet, increased the usable x-ray flux on the sample by three orders of magnitude compared to typical (kHz) synchrotron-based time-resolved experiments. This led to XES data quality fully comparable to that of steady-state experiments, but now with sub-nanosecond time resolution.
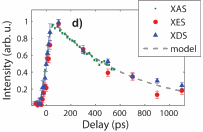
The increase in flux and the experimental design also allowed simultaneous collection of high-quality scattering data in only a few hours, despite the relatively low scattering power of the solute and the disordered solvent. By combining the XES data with the XDS results, the presence of the high-spin configuration of the FeII ion could be unambiguously tied to the global structural changes in molecular structure (Fig. 2).
The superior signal-to-noise ratio of the scattering data combined with the comparatively gentle laser excitation further allowed the researchers to tentatively confirm a key recent theoretical prediction regarding photo-induced solvation changes around [Fe(bpy)3]2+. The solvation dynamics of HS [Fe(bpy)3]2+ following photoexcitation were seen to lead to a transient increase in solvent density with a time evolution closely matching that of the solute. The observed 0.1% increase in density is in agreement with the theoretical prediction that approximately two water molecules are expelled from the solvation shell due to the structural and electronic changes caused by the photoexcitation.
Even though [Fe(bpy)3]2+ is a thoroughly studied model compound in time-resolved science, this experimental detection of a process involving only the disordered rearrangement of light elements highlights the extent to which increased x-ray flux will allow future time-resolved experiments to move on to classes of systems that have so far only been studied with steady-state methods. — Vic Comello
See: K. Haldrup*1, G. Vankó**2, W. Gawelda3, A. Galler3, G. Doumy4, A. M. March4, E. P. Kanter4, A. Bordage2, A. Dohn5, T. B. van Driel1, K. S. Kjær6, H. T. Lemke7, S. E. Canton8, J. Uhlig8†, V. Sundström8, L. Young4, S. H. Southworth4, M. M. Nielsen1, and C. Bressler3, “Guest–Host Interactions Investigated by Time-Resolved X-ray Spectroscopies and Scattering at MHz Rates: Solvation Dynamics and Photoinduced Spin Transition in Aqueous Fe(bipy)32+,” J. Phys. Chem. A 116, 9878 (2012). DOI:10.1021/jp306917x
Affiliations: 1Technical University of Denmark, 2Hungarian Academy of Sciences, 3European XFEL, 4Argonne National Laboratory, 5Danish Technical University, 6University of Copenhagen, 7SLAC National Accelerator Laboratory, 8Lund University. †Additional address: National Institute of Standards and Technology
Correspondence: *[email protected], **[email protected]
This project was supported by the European Research Council via Contract ERC-StG-259709, the Danish National Research Foundation's Centre for Molecular Movies, the European XFEL, and DANSCATT. A.M.M., G.D., S.H.S., E.P.K., and L.Y. acknowledge support from the U.S. Department of Energy (DOE) Office of Science, Division of Chemical, Geological and Biological Sciences, under Contract No. DE-AC02-06CH11357. K.H. gratefully acknowledges support from the Carlsberg and Villum Foundations. G.V. acknowledges support from the Bolyai János Fellowship of the Hungarian Academy of Sciences. V.S. acknowledges support from the European Research Council, Advanced Investigator Grant, VISCHEM-226136. S.E.C. gratefully acknowledges funding from the Swedish Research Council. C.B., A.G. and W.G. acknowledge funding from the German Research Association DFG via SFB925 (project A4). Use of the Advanced Photon Source, an Office of Science User Facility operated for U.S. DOE Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357.
The Advanced Photon Source at Argonne National Laboratory is one of five national synchrotron radiation light sources supported by the U.S. Department of Energy’s Office of Science to carry out applied and basic research to understand, predict, and ultimately control matter and energy at the electronic, atomic, and molecular levels, provide the foundations for new energy technologies, and support DOE missions in energy, environment, and national security. To learn more about the Office of Science x-ray user facilities, visit http://science.energy.gov/user-facilities/basic-energy-sciences/.
Argonne National Laboratory seeks solutions to pressing national problems in science and technology. The nation's first national laboratory, Argonne conducts leading-edge basic and applied scientific research in virtually every scientific discipline. Argonne researchers work closely with researchers from hundreds of companies, universities, and federal, state and municipal agencies to help them solve their specific problems, advance America's scientific leadership and prepare the nation for a better future. With employees from more than 60 nations, Argonne is managed by UChicago Argonne, LLC for the U.S. Department of Energy's Office of Science.